Do we accept that biological phenomena are unfathomable?
Human understanding of the world around us is at risk. Advances in AI and machine learning, in parallel with exponential increases in the accumulation of data and computer power, are stagnating our ability to make sense of natural processes.
Drug discovery is a field with such an increasingly constipated outlook. And the consequences of “black-box medicine” (defined as: “the use of opaque computational models to make decisions related to health care” [Nicholson Price II, 2015 ]), could be dire.
Machine learning in medicine.
Machine learning increasingly relies on black-box models, the workings of which are not humanly comprehensible. These opaque computational models find patterns in data to predict some given outcome of interest. Within medicine, such outcomes could be a diagnosis, an optimised drug design, or a patient’s risk of re-hospitilization.
This means that the users applying these predictions are not served any explanation as to how the model came up with a given prediction, and the same goes for the patient whose treatment is determined by such a model. These models are chosen based only on their predictive performance due to a lack of alternatives that actually could provide transparency.
The increasing application of black-box models gives rise to a range of both ethical and scientific questions.
Legally, clinicians are to blame for any wrongdoings in the clinic, which also goes for any clinical decision taken with support from a software programme, including machine learning-based software. For that reason — as either a person giving or receiving care — you need to have confidence that the clinician can understand and explain how a machine learning model works.
Simply put: The clinician should trust the model in order to apply it. The odds are they cannot — until recently.
The alternative is transparency.
There is a lack of access to the actual patterns within data when using black-box algorithms. Consequently, models act more like passive filtering tools than active enablers for the scientists responsible for designing new, innovative medicines.
But with a transparent and interpretable model framework, scientists can disentangle the massive amount of data gathered to discover new mechanisms leading to improved medicines.
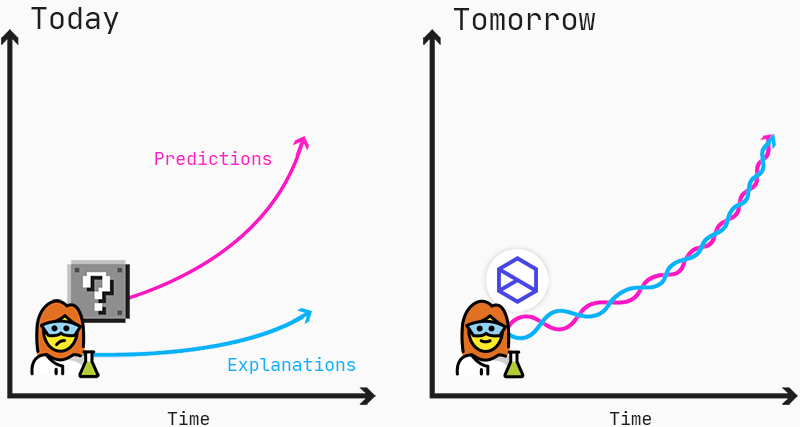
Predictions and explanations should go hand-in-hand to enable scientists to find new insights into mechanisms of diseases and drug responses.
Duchenne muscular dystrophy.
We want to improve the treatment of Duchenne muscular dystrophy (DMD), a neuromuscular disorder that affects 1:5,000–10,000 male births. DMD is caused by mutations in the DMD gene, which codes for the dystrophin protein. Dystrophin is an important structural protein in muscle cells, and a lack of dystrophin results in progressive muscle weakness which typically begins before the age of 6, followed by the inability to walk by the age of 12. Death usually occurs in the second decade of life, often resulting from heart failure [Kieny et al, 2013].
Antisense Oligonucleotides.
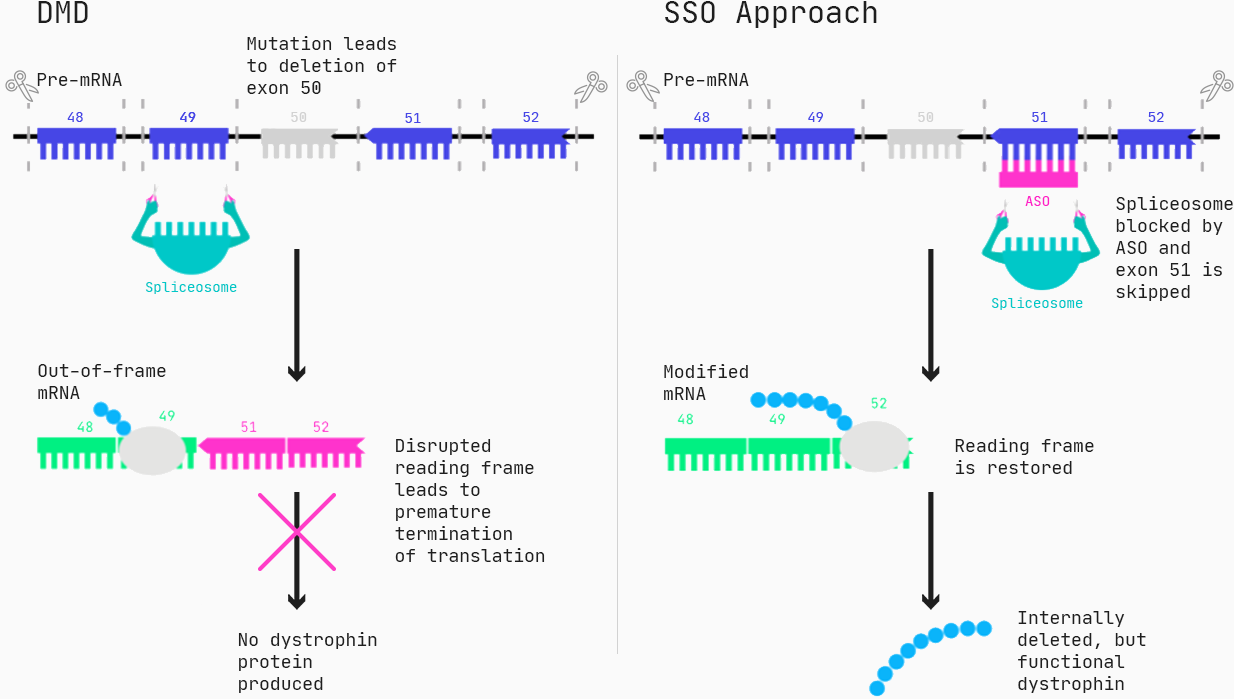
Approximately 60–70% of the patients suffering from DMD have mutations that lead to deletions of one or more exons (generally, an exon is the part of a gene that gets expressed as protein), resulting in a malfunction of the dystrophin protein.
One type of drug that can restore the function of dystrophin for these patients are Antisense Oligonucleotides (ASOs), specifically in the form of splice switching ASOs. Via Watson-Crick pairing, the ASOs bind to the target, and for splice-switching ASOs, this can cause exons to be incorrectly included or excluded, essentially changing the protein produced from a gene.
As for all drug modalities, ASO molecules have a series of properties such as toxicity, efficacy, and cellular uptake, all relevant for ultimately designing an effective drug. You want to make a safe drug, you want to make a drug that is effective, and you want to make a drug that reaches its destination in the body — so how do we know which ASO molecules are non-toxic and work?
In the case of DMD, we know up-front which sequences to target. But from then on, we test hundreds to thousands of molecules in example cells to pick out those with the right properties. Having measured the efficacy of all these molecules, we would like to be smarter for the next round of molecule design. Both to increase the hit rate and to learn more about how splice-switching ASOs work.
Annotating molecules.
The only limit to annotating ASOs and extract information from molecules is our creativity. This is due to the string-like nature of these molecules and the many ways to describe how they bind to their target. How do we then select the features that are most related to a certain drug property? How do these features combine to make a molecule effective? And where can we do more research to increase the likelihood of discovering a molecule with the right drug properties? How does the model outcome change as we change the model input?
To answer these questions, we must prioritize understanding the underlying mechanisms instead of considering performance metrics only.
Introducing the model framework to the QLattice — a high-performing and explainable AI — gives us an arena to narrow down on a few driving features. This tool helps us explore how features relate in predicting the outcomes. The only requirement is that data is in a tabular format.
Data.
All data and features presented here are taken from the paper: Annemieke Aartsma-Rus, et al. “Guidelines for Antisense Oligonucleotide Design and Insight Into Splice-modulating Mechanisms” Molecular Therapy (2009).
The published data contains 156 splice switching oligonucleotides (SSOs) previously designed and evaluated for splice modulation of the dystrophin transcript. The SSOs target different exons of the DMD gene, and a SSO is classified as effective if the exon-skipping is at least 5%.
Features.
Each SSO is annotated with 42 different features. A few examples are:
Number and percentage of A, C, U, and G nucleotides.
Predicted exonic splicing enhancer (ESE) sites that facilitate splicing by binding of so-called Ser-Arg-rich (SR) proteins.
The minimum free energy for the secondary structure of the SSO and the duplex formed between identical SSOs.
Setting up Python and reading in data.
Split in train and test.
We train on our training set and evaluate on our test set (in this demo, we leave out the validation set).
The QLattice application.
We connect to our QLattice and define the semantic types of our features (categorical or numerical).
Model search.
Now we search across all 42 features with a wide set of available mathematical operations to point to the simplest models that explain efficiency in SSOs. The architecture of the models is “set free”, i.e. all sorts of interactions and non-linear relationships are in scope.
Ultimately, we are looking to condense phenomena to the simplest possible form without going too simple!
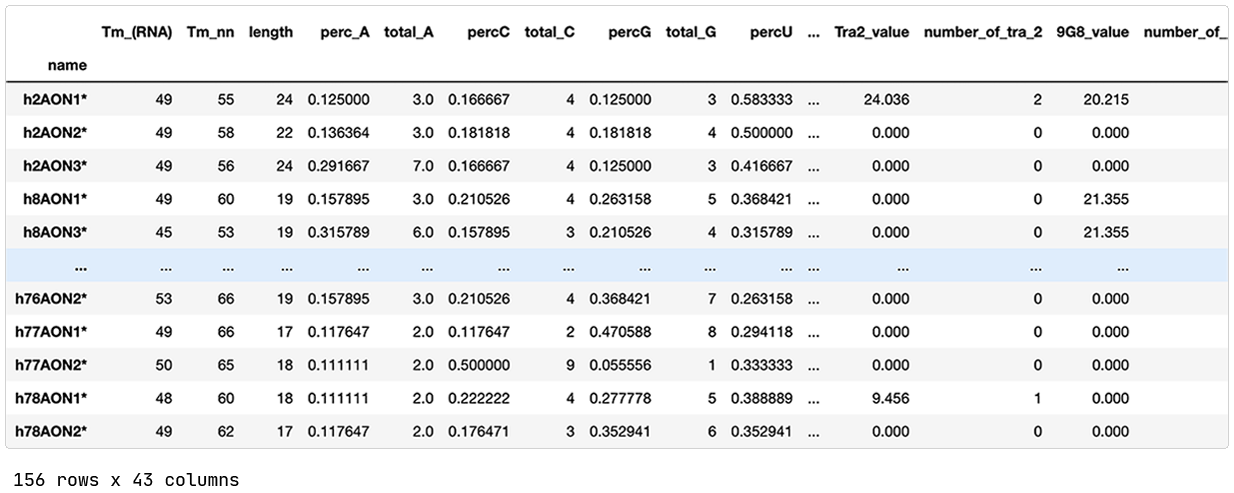
Pick up the preferred model and inspect.
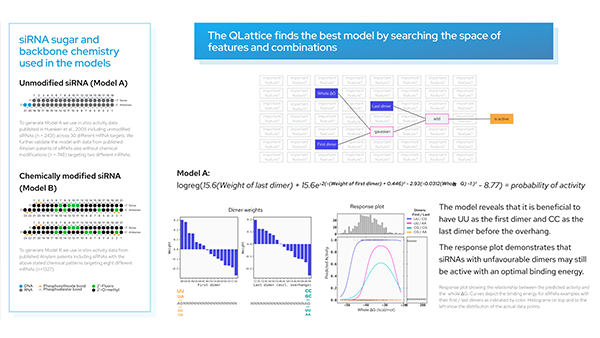
We have a list of models sorted by ‘BIC’ (Bayesian information criterion) — a criterion that helps the QLattice balance performance with complexity. The best-performing model includes three features; ‘delta_Energy_AON_target_binding’, ‘RescueESE_hexamers’, and ‘Tra2_value’.
The features are added together to ultimately deliver a set of predicted probabilities of SSO efficiency. Since we are solely doing ‘add’ operations to our inputs, we have found a linear model.
Math.

These models are just math and thus can be expressed as mathematical formulas. Our chosen model is shown above.
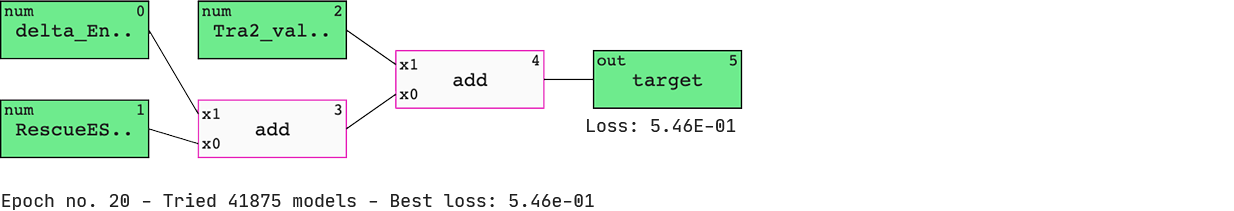
Signal flow.
To understand the origin and flow of signal in the model, we colour it by Pearson correlation. Also, we display the standard metrics of a binary classification case for both the train and test set.
This model generalises well to new data; the AUC of the train and test set are similar. This means that if we were to be served with a 50%-50% chance of an efficient or non-efficient SSO, we would have a 75% chance of predicting the right outcome.
Interpreting the model.
The above model points us to three features that are important for designing efficient SSOs:
- High numbers of rescueESE hexamers
- Low values of Tra2β
- High delta energy between ASO and target
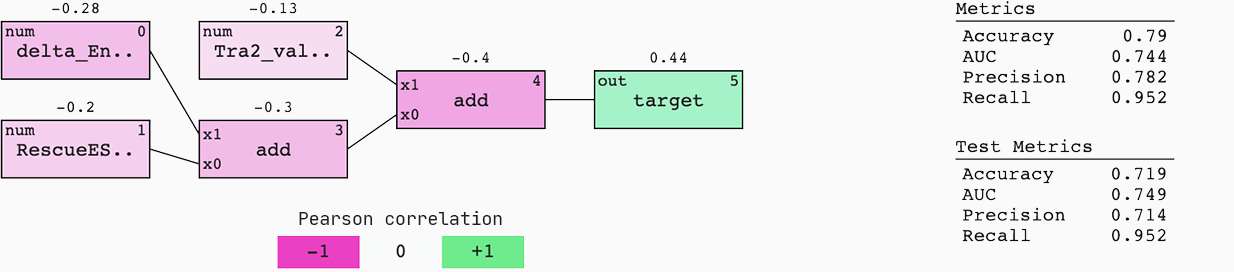
This is evident both from reading the mathematical formula but it can also be found by applying a set of plots explaining the model dynamics. Let’s investigate how these features depend on each other.
In the partial plot below, we are looking at our model’s predictions when we let the binding energy between ASO and target vary for a fixed set of values for ‘Tra2-value’ and ‘RescueESE_hexamers’. It becomes obvious that higher binding energy leads to a higher probability of an efficient SSO. This is most pronounced when we see a lower number of Tra2-values combined with a higher number of hexamers (the teal line), whereas for higher numbers of Tra2-values and a lower number of hexamers (pink line), we need higher binding energy to raise the probability of seeing an efficient SSO.
Conclusion.
So what just happened?
In a few lines of code, we found the best-suited model to explain the efficiency of SSOs based on a set of 42 related features. The model is transparent by design since every operation is given by a mathematical formula, and the workings of the model can be dissected by various visualisations (most of which are not presented in this blog post).
We found a model that can help drug discovery researchers in classifying the efficiency of new SSO drug candidates. And not only that, we also sparked the interest of the researchers to ask why these three features are strongly associated with efficiency in this way. Hopefully, new and exciting research questions emerged. The black-box counterpart to this model would have given a set of typically equally good predictions but left the researchers in the dark about how and why.
What a bummer that science could have ended right there, just when things started getting exciting. Thanks, QLattice!
Thanks for reading, and if you’re inspired, feel free to reach out to the Abzu team!
References:
Kieny P et al. “Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper centre between 1981 and 2011”. Ann Phys Rehabil Med (2013)
W. Nicholson Price II. “Black-box medicine”. Harvard Journal of Law & Technology (2015)