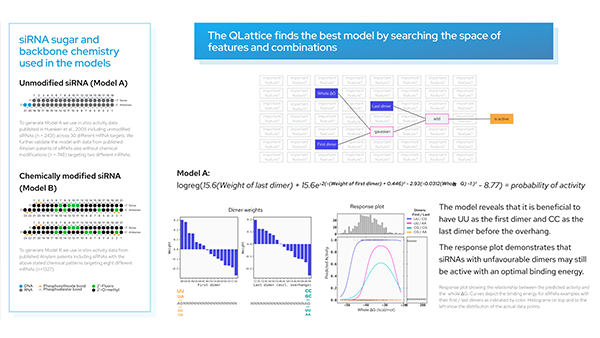
The conventional methods of cancer treatment, chemotherapy and radiotherapy are still the main methods of treatment against cancer, although they have devastating side effects [1]. A limitation is that they kill not only cancerous cells but also the cells around them, causing hair loss, pain, fatigue and nausea [1].
New treatments are being developed that specifically target cancer cells. One example is peptides.
Peptides are typically less toxic because they are easily broken down into their building blocks (amino acids) by the body’s own enzymes. However, a difficulty in developing peptide treatments is that they require significant R&D investment to improve target efficiency and reduce production costs [2].
Therefore, we will use the QLattice® to build an explainable AI model to help us understand the combined properties of a better anti-cancer peptide. Symbolic regression models are quite precise in searching for and selecting relevant chemical and structural properties of peptides so that existing drugs can be further developed.
Additionally, bringing as much disease understanding and drug design work in silico will reduce the cost of peptide drug development and increase the speed to market.
Let’s go!
Peptide drug development: The details.
Peptides with specific chemical properties have been shown to be toxic to bacteria due to the negative charge on the cell surface. Charged peptides are able to interact with and destroy the membrane, causing cells to break apart [1], often referred to as cell lysis. Peptides are therefore a promising cancer therapy because cancer cells share the same electrochemical properties, having a charge distributed over their entire surface [1].
We can address these therapeutic possibilities by calculating and analyzing different chemical properties of anti-cancer peptides with the aim of further modifying the peptides so that they can lyse cells even better.
Features for peptide drug development.
A number of features of the chemical and physical properties, such as hydrophobicity, polarity, and charge, can be calculated from the amino acid sequence. These will serve as inputs into the QLattice.
With this, we create a classification model that determines whether the peptide is an anticancer peptide (1) or not (0). This will give us insights into what a good anticancer peptide should (or should not) look like.
For this example, we’ll be using a publicly available dataset of annotated peptides published in the paper ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation [4].
Building the model.
We calculated a set of chemical and structural peptide features: Alpha-helix propensity, beta-sheet propensity, coil propensity, aromaticity, molecular weight (mw), flexibility, instability, half-life, hydrophobicity, extinction coefficient reduced and oxidised, isoelectric point, and sequence length.
In addition, fractions of all 20 amino acids and amino acid pairs such as KC (lysine followed by cystine) and FL (phenylalanine followed by leucine) were considered as input data.
To adequately capture the hydrophobicity of peptides, a biological scale for hydrophobicity was chosen that captures the ability of a sequence to be incorporated into the lipid membrane.
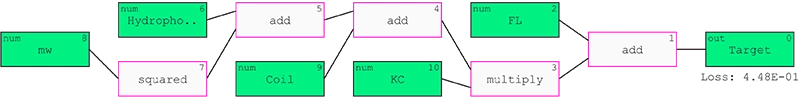
In total, 434 data columns were used from which the QLattice was set out to select the most important. From all the hypotheses generated, one hypothesis was selected that contained 5 features hypothesised to be relevant to membrane interaction: Molecular weight, hydrophobicity, coil propensity, and FL and KC composition.
The selected model predicted the target with an accuracy of 0.798 and an AUC of 0.869 on the test data (Figure 1). The confusion matrix in Figure 2 visualises the performance by showing the expected predictions relative to the actual predictions.
Figure 1: QLattice model predicting target outcome.
The QLattice model predicts the target outcome with an accuracy of 0.799, an AUC of 0.869, a precision of 0.788, and a recall of 0.82 on the test data.
Figure 2: Confusion matrix.
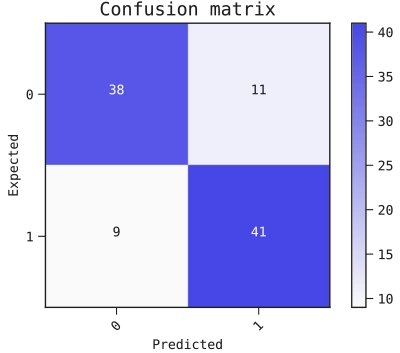
The confusion matrix rates our performance by showing the expected predictions relative to the actual predictions. This confusion matrix has a threshold of 0.5 for the predictions made from the test data.
Peptide drug development: Understanding the model.
The selected model captured several chemical/structural properties of peptides that have been shown to determine the anti-cancer properties.
As expected, hydrophobicity and coil expansion play an important role in the peptide membrane interaction. This can be understood as “hydrophobicity describes the potential of a peptide to interact with the membrane and trigger lysis”.
Furthermore, it is known that hydrophobic coil regions can intercalate into the lipid membrane. Interestingly, peptides with a high potential to insert into the membrane showed a low possibility of destroying cancer cells. Moreover, peptides with low coil propensity in the lower hydrophobic regions were found to be effective in lysing cancer cells (Figure 3).
Figure 3: Coil propensity and hydrophobicity plot.
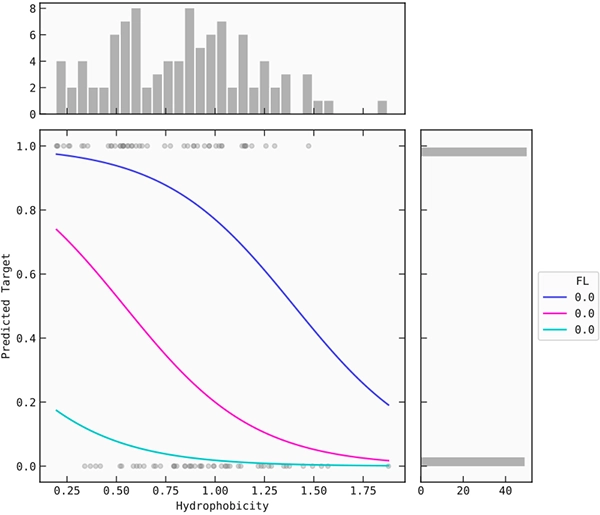
By setting the molecular weight and the proportion of FL and KC to a fixed value (median), you can plot the coil propensity and hydrophobicity by their signal against the target. Three different values of coil propensity are plotted against hydrophobicity.
Peptide drug development learnings:
From this, we have learned that peptides with low membrane penetration possibilities, reflected in secondary structure and biological hydrophobicity, are promising in the development of anticancer peptide drugs.
Additionally: The presence of the KC within a sequence (one very polar and one charged amino acid) proved particularly relevant for anti-cancer properties. The target was predicted to be an anticancer peptide for all peptides with KC present.
Cysteine, in particular, was shown to lead to cell-penetrating and antimicrobial activity of a peptide sequence. Its cell-destructive properties have already been observed in the snake venom crotamine, which is rich in cysteine residues [5].
From insights to action.
We have now learned about some properties we should focus on for anti-cancer peptides: Hydrophobicity, KC composition, and alpha-helical regions.
We focused on exploring the specific mechanism of the therapeutics themselves: The attack on the membrane. Introducing KC amino acid pairs into a sequence leads to a higher lysis ability of cancer cells.
This example shows that there are opportunities to use symbolic regression to find properties and amino acids that can be used in modifying peptide sequences at the early stages of drug development. Two major challenges lie ahead: Peptides have a short biological half-life and penetrate poorly into tissues [2][3].
The analysis in this blog post can be applied to these two problems to gain insights into what influences toxicity, solubility, and stability.
References:
[1] Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta. 2008 Feb;1778(2):357-75. doi: 10.1016/j.bbamem.2007.11.008. Epub 2007 Nov 22. PMID: 18078805; PMCID: PMC2238813.
[2] https://www.prnewswire.com/news-releases/global-peptide-cancer-therapeutics-market-2021-to-2026—drug-dosage-price–clinical-trials-insight-301248412.html [2022-02-18]
[3] David J. Craik & Meng-Wei Kan (2021) How can we improve peptide drug discovery? Learning from the past, Expert Opinion on Drug Discovery, 16:12, 1399-1402, DOI: 10.1080/17460441.2021.1961740
[4] Yi H-C, You Z-H, Zhou X, Cheng L, Li X, Jiang T-H, et al. ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation. Molecular Therapy – Nucleic Acids. 2019;17:1-9. doi: 10.1016/j.omtn.2019.04.025.
[5] Ponnappan, Nisha, Deepthi Poornima Budagavi, and Archana Chugh. “CyLoP-1: Membrane-active peptide with cell-penetrating and antimicrobial properties.” Biochimica et Biophysica Acta (BBA)-Biomembranes 1859.2 (2017): 167-176.